
Home
Welcome!
Our research lies at the interface of chemistry, biology, and genomics
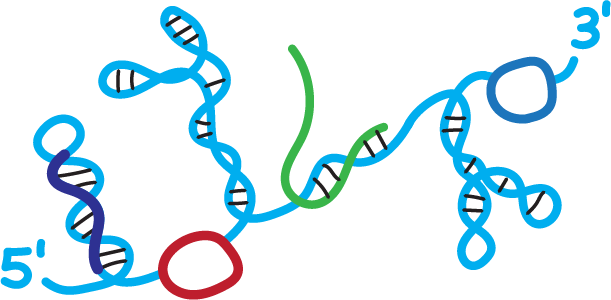 We create chemistry-based technologies, or “molecular microscopes”, that reveal the structure of large RNAs in detail, in viruses and cells. Our lab then leads efforts to understand how RNA structure regulates diverse areas of biology and to advance the fast-evolving field of RNA-targeted drug discovery. Work in our lab is driven by a fundamental attitude: We try to make technologies simple. Making things simple is hard but, when you are successful, simplicity builds in robustness and inspires others to adopt and extend our foundational work.
We create chemistry-based technologies, or “molecular microscopes”, that reveal the structure of large RNAs in detail, in viruses and cells. Our lab then leads efforts to understand how RNA structure regulates diverse areas of biology and to advance the fast-evolving field of RNA-targeted drug discovery. Work in our lab is driven by a fundamental attitude: We try to make technologies simple. Making things simple is hard but, when you are successful, simplicity builds in robustness and inspires others to adopt and extend our foundational work.
One of the most amazing discoveries of recent years has been a growing understanding of the profound roles of RNA in regulating all areas of biology. The functions of all RNA molecules are deeply influenced by two features. First, the sequence, which is now easy to determine and, second, by the ubiquitous and often complex structures that form when a long RNA folds back on itself. Until relatively recently, however, we had little idea of the broad interrelationships between RNA structure and function because there simply did not exist accurate strategies for examining the large-scale architecture of RNA molecules.
Impact
 Our lab addresses this knowledge gap – of simply not knowing much about the structure of RNAs in cells – by inventing and applying quantitative and high-throughput chemistry-based strategies to interrogate RNA structure. Technologies including SHAPE, mutational profiling (MaP), and single-molecule correlated chemical probing (smCCP) are now in use worldwide and are transforming our understanding of how RNA structure governs biological regulatory networks.
Our lab addresses this knowledge gap – of simply not knowing much about the structure of RNAs in cells – by inventing and applying quantitative and high-throughput chemistry-based strategies to interrogate RNA structure. Technologies including SHAPE, mutational profiling (MaP), and single-molecule correlated chemical probing (smCCP) are now in use worldwide and are transforming our understanding of how RNA structure governs biological regulatory networks.
The most critical insight obtained through application of our technologies is that the pattern of RNA base pairing (or secondary structure) functions broadly as a switch that enables or modulates ubiquitous classes of RNA-centered gene regulation. Diverse biotechnology companies interested in targeting RNA rely on technologies invented in our lab to identify ligand-binding RNA structures and to advance small molecules into drugs.
What’s Next
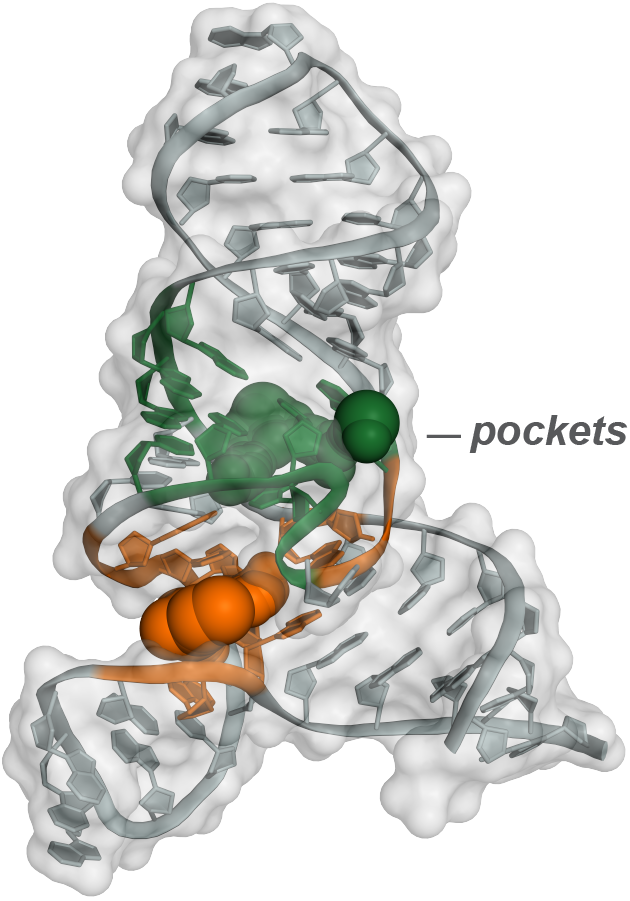 The next frontier in large-scale RNA structure analysis lies in understanding the pervasiveness and functions of higher order tertiary structures. Our laboratory is inventing multiple – ultimately simple (!) – technologies to address this challenge. RNA motifs with complex higher order structure are exactly the places most likely to contain clefts and pockets that are targetable with function-modifying small molecules. Our efforts in the emerging field of RNA-directed drug discovery support a new way of thinking about drugging RNA and are poised to transform broad therapeutic areas by targeting currently ‘undruggable’ processes.
The next frontier in large-scale RNA structure analysis lies in understanding the pervasiveness and functions of higher order tertiary structures. Our laboratory is inventing multiple – ultimately simple (!) – technologies to address this challenge. RNA motifs with complex higher order structure are exactly the places most likely to contain clefts and pockets that are targetable with function-modifying small molecules. Our efforts in the emerging field of RNA-directed drug discovery support a new way of thinking about drugging RNA and are poised to transform broad therapeutic areas by targeting currently ‘undruggable’ processes.